

Commentary by:
Contributors
Assessing the Heterogeneity of Single Cancer Cells using Primary Template directed Amplification (PTA)
Primary Template-directed Amplification (PTA) is a novel and accurate whole-genome amplification (WGA) method for the genomic analysis of single cells. Here we demonstrate the importance of understanding the heterogeneity of genomic modifications that occur between individual cells within a cancer specimen.

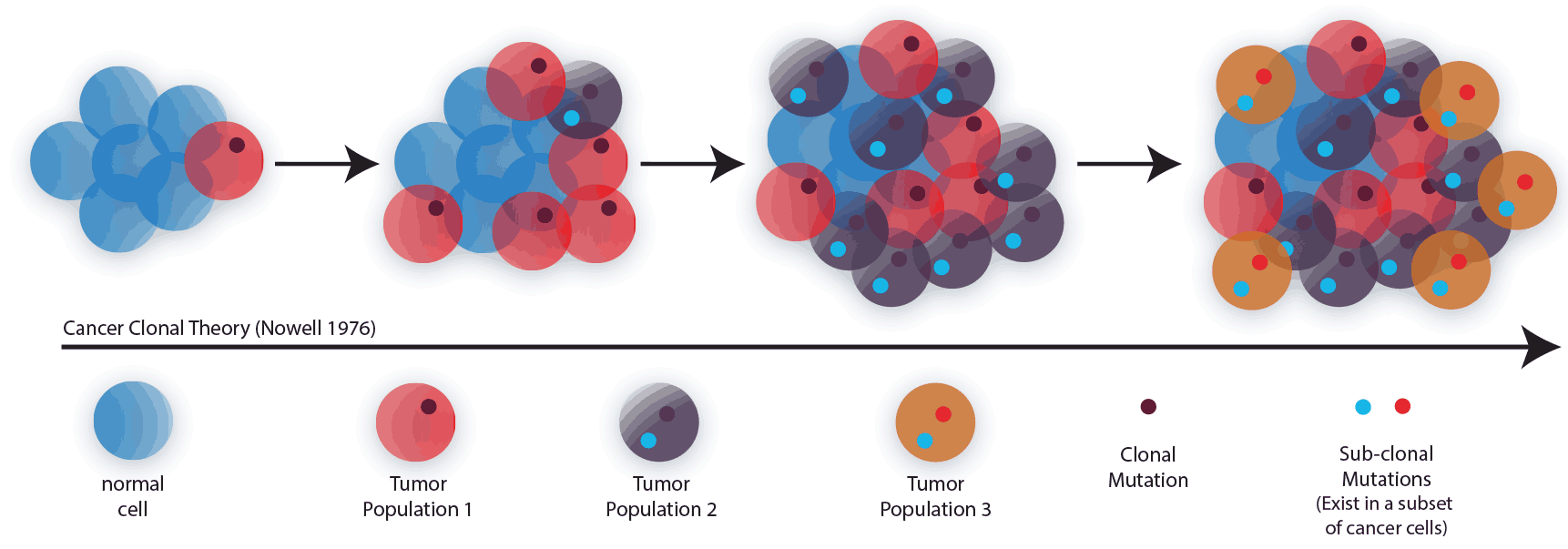
Figure 1. Clonal Theory of Cancer.
Introduction
In 1976 the clonal theory of cancer was published (Figure 1) by Peter Nowell1. Since this time, forty-five years ago, a tremendous effort has been put forth to determine the molecular basis of cancer through a variety of technical methods2. However, despite the myriad of methods employed to study the disease, there has been a lack of tools to determine the most basic element of oncology, the individual cells that allow the disease to spread and adapt. We posit that to improve outcomes for patients, one must be able to decipher the genetics of the disease within these pathogenic cells. Cancer is a complex disease. While it may in fact arise from a single cell, the diseased cells are under constant evolutionary pressure. As a result, several aspects of the pathology are important to consider.
- Cancer is not a monolithic disease, the heterogeneity of the cells allows the disease to progress and adapt.
- Cancer is not defined by a small subset of genes. As detection of variation becomes more sensitive so will the number of associated genes and variants within a pathology.
- Mutation co-occurrence drives disease. Individual cells or clones harbor specific combinations of gene variants which escalate the disease to become pathogenic and persistent.
- Single cell tumor mutation burden (TMB) contributes to the evolutionary path of the disease and may reveal how to target it for destruction.
- A combination of changes in the genome drive the functional status of the cell, including copy number variation (CNV), structural variation and single nucleotide variation (SNV).

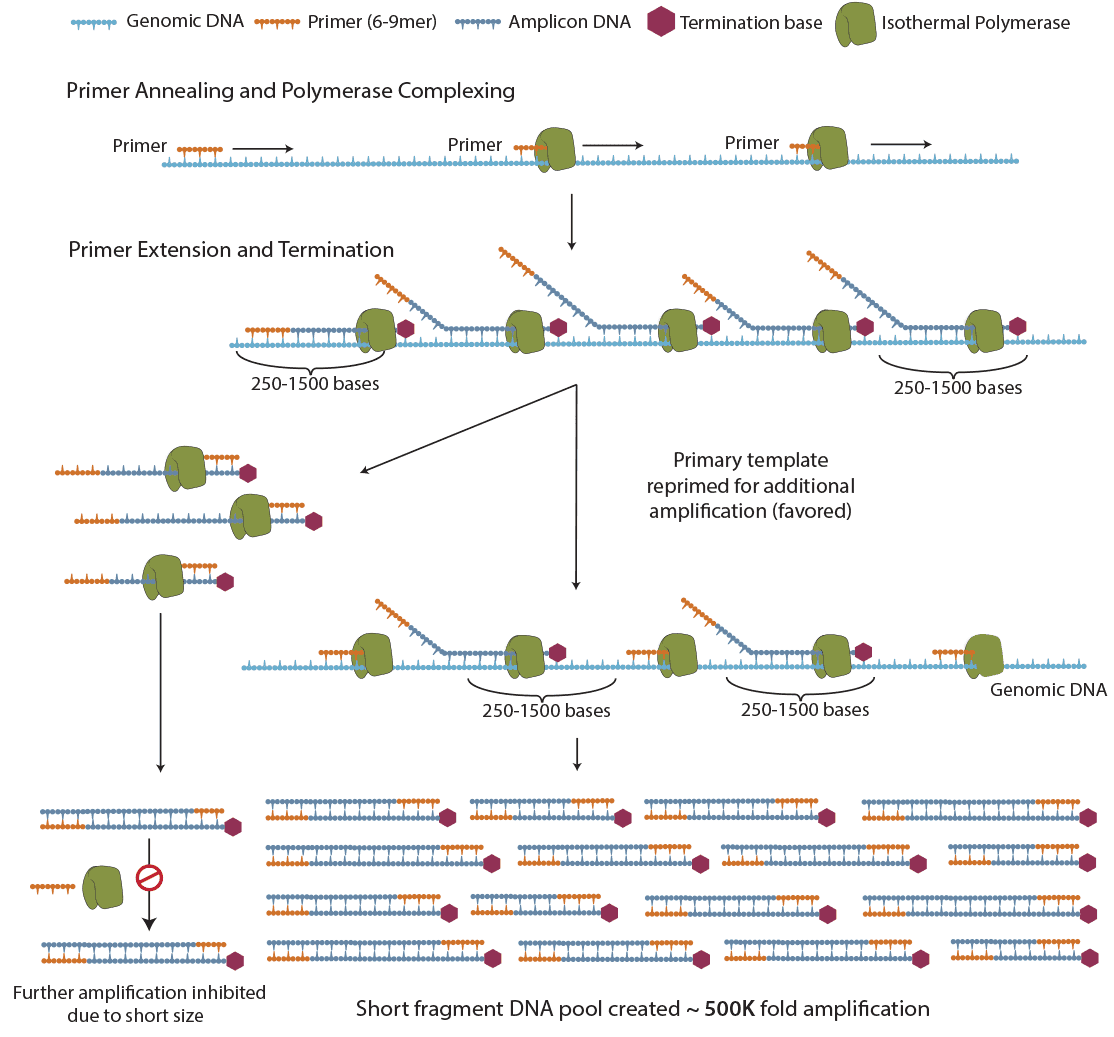
Figure 2. The principle of PTA. ResolveDNA® provides unbiased amplification by utilizing random priming combined with novel termination chemistry to produce a true representation of original sample template. The use of proprietary nucleotides prevents the production of long amplicons, which are kinetically unfavored to be re-copied during the amplification reaction. By limiting the size of the produced amplicon, primers are re-directed to the primary template.
The significance of PTA
Due to the complexity of cancer there is no simple diagnostic or therapeutic solution. While the modalities of analysis, (i.e. transcriptome, methylome, proteome) may reveal global influences on the evolution of cancer, the genome is the blueprint and a key to understanding how individual cells dictate patient outcomes. To understand the complexity of a tumor, one needs to sequence individual cells from the tumor's cellular population. In order to sequence a genome from a single cell, its DNA needs to undergo whole genome amplification(WGA) to have sufficient quantities of DNA for WGS. Primary Template directed Amplification (PTA) was designed to solve this challenge (Figure 2). PTA is a novel, isothermal method that overcomes the challenges associated with single cell WGA, such as low and/or uneven genome coverage, allelic skewing or dropout, and experimental artifacts3. Using PTA we are now able to reveal these rare single cell alterations that exist, persist and expand during the cancer life-cycle.
Using a simple step-wise protocol, PTA4 allows the amplification of the entire genome from an individual cancer cell. The process generates the highest uniformity of amplification reported in the literature for low inputs of DNA and single cells. Combining PTA with the enrichment power of FACS then enables the enrichment of these lethal cells and the ability to decipher the genomic mechanisms allowing them to survive, even thrive, under therapeutic pressure.
Cancer is not a monolithic disease. Cellular heterogeneity allows the disease to progress and adapt
The development of next generation sequencing (NGS) has enabled the scientists in the field of oncology to characterize the most abundant mutations present in tumors5. However, the analysis of tumors as an aggregate, assumes the disease is monolithic.

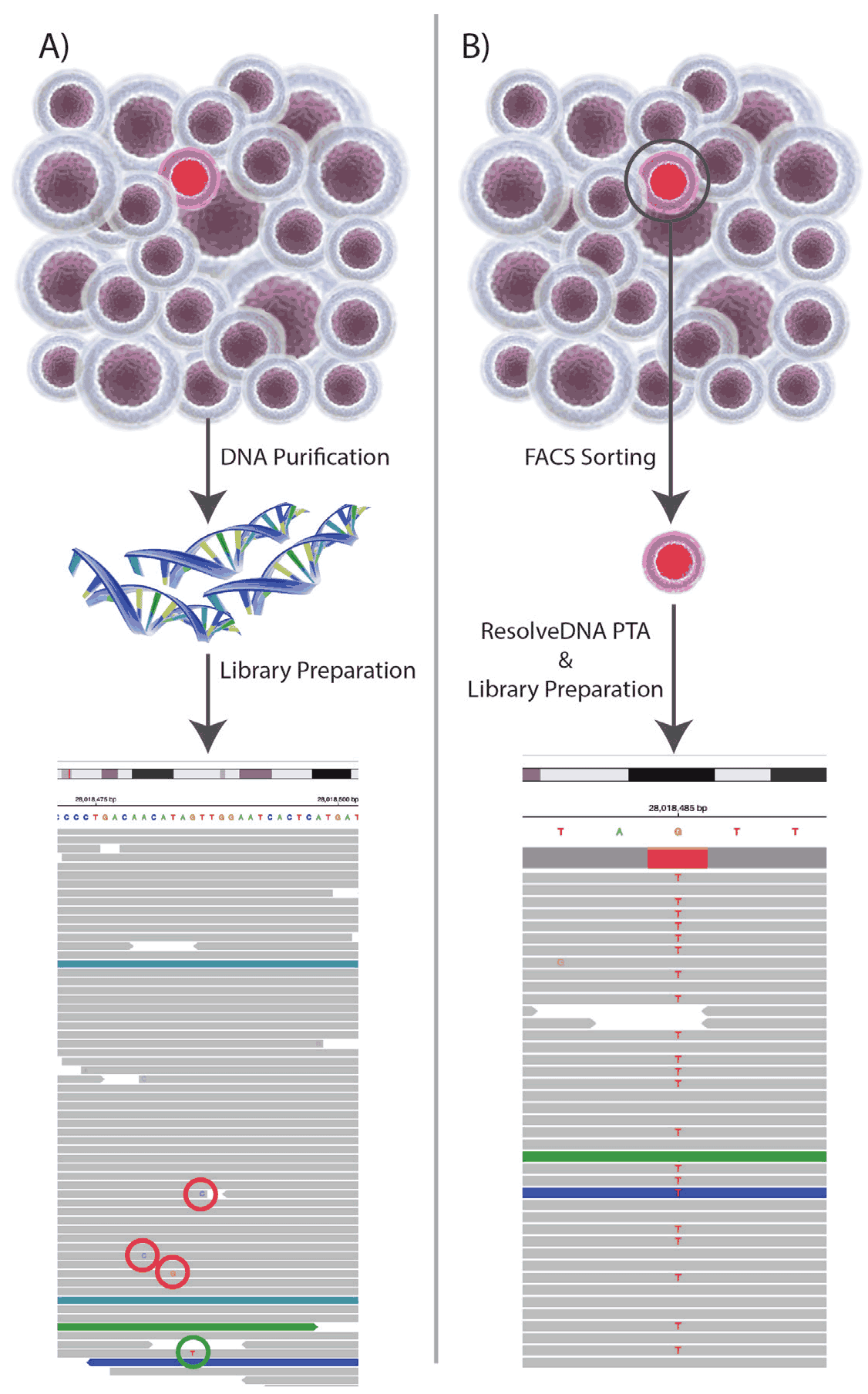
Figure 3. The Barnyard Experiment: 50 cells (49 GM12878 + 1 Molm13 AML cell) were analyzed as either a bulk sample or as individual cells. The data reveal the limitations of detecting a variant allele within a bulk DNA sample(A) from a population of cells, where the divergent mutation (green circle) is only seen in one read within ~ 40 reads which cannot be differentiated from sequencing error (red circles). Alternatively, when cells are analyzed individually, the variant somatic allele is detected at ~ 50% of reads in each cell (B). VAF then becomes a function of how many cells are analyzed. In the current example, this translates to (1cell/50cells)*100)/2copies of the genome = 1.0%. As the numbers of cells increase to ~ 384, projected VAF is ~ .13% or roughly a 40 fold improvement in sensitivity over the most popularly utilized conventional bulk sequencing approaches.
One tumor may have hundreds of genomes. This complexity or heterogeneity at the genome level is defined as variant or minor allele frequency (VAF/MAF) or the ability to detect specific alterations in a specific cell. The analysis of single cell genomes provides unparalleled VAF sensitivity and specificity compared to existing research and diagnostic solutions. To illustrate this concept, we have recently conducted a simple experiment to demonstrate the ability to increase VAF sensitivity using the ResolveDNA single cell amplification platform.
The "Barnyard" experiment, mixing cellular species, demonstrates that sequencing of single cells can reveal not only the most prevalent mutations, but those mutations that are more rare, which drive the long-term progression of cancer. DNA extracted from tumors for NGS analysis can often originate from samples of million of cells. However, sequencing analysis is limited to 35X for WGS, 100X for WES, ~ 2000X for a targeted panel. Which means, for a sample with 20% tumor burden there is ~10% probability to detect the variants in these cells. This is only for the most dominant variants, those with a specific variant in every cell. By contrast, single cell sequencing using PTA allows for the detection of rare variants, the sensitivity of which is only limited by the number of cells analyzed in a given sample.
To illustrate this we provide a prophetic example (Figure 3) where 1200 cells from a tumor are collected and homogenized as a group and further sequenced. A sample containing 20% tumor generates approximately 3 complete reads when sequenced at 40X (Figure 3B). If a secondary mutation presents at a lower frequency (i.e. 2%) it is estimated to have zero complete reads for the tumor variant in the sample (Figure 3B). This is due to the overwhelming presence of "normal and stromal" DNA that dominate the sample. The detection of these rare variants is further complicated by the presence of sequencing errors that occur in NGS analysis between 0.1-1%. Overall, sequencing of cell homogenates is severely limited to only the most prevalent mutations and in many cases does not facilitate the eradication of the disease, which may result in relapse.
Alternatively, if the tumor cells are singulated and analyzed individually three main patterns emerge; those that are normal, the tumor variant cells, and the tumor cells containing a secondary variant allele (Figure 4). Often it is tumor secondary variant alleles that drive the failure of therapeutics6. Detection of these secondary variant alleles is made possible by the single cell read structure. When analyzing a variant allele, one of three possibilities exist (Figure 4E), the absence of a variant

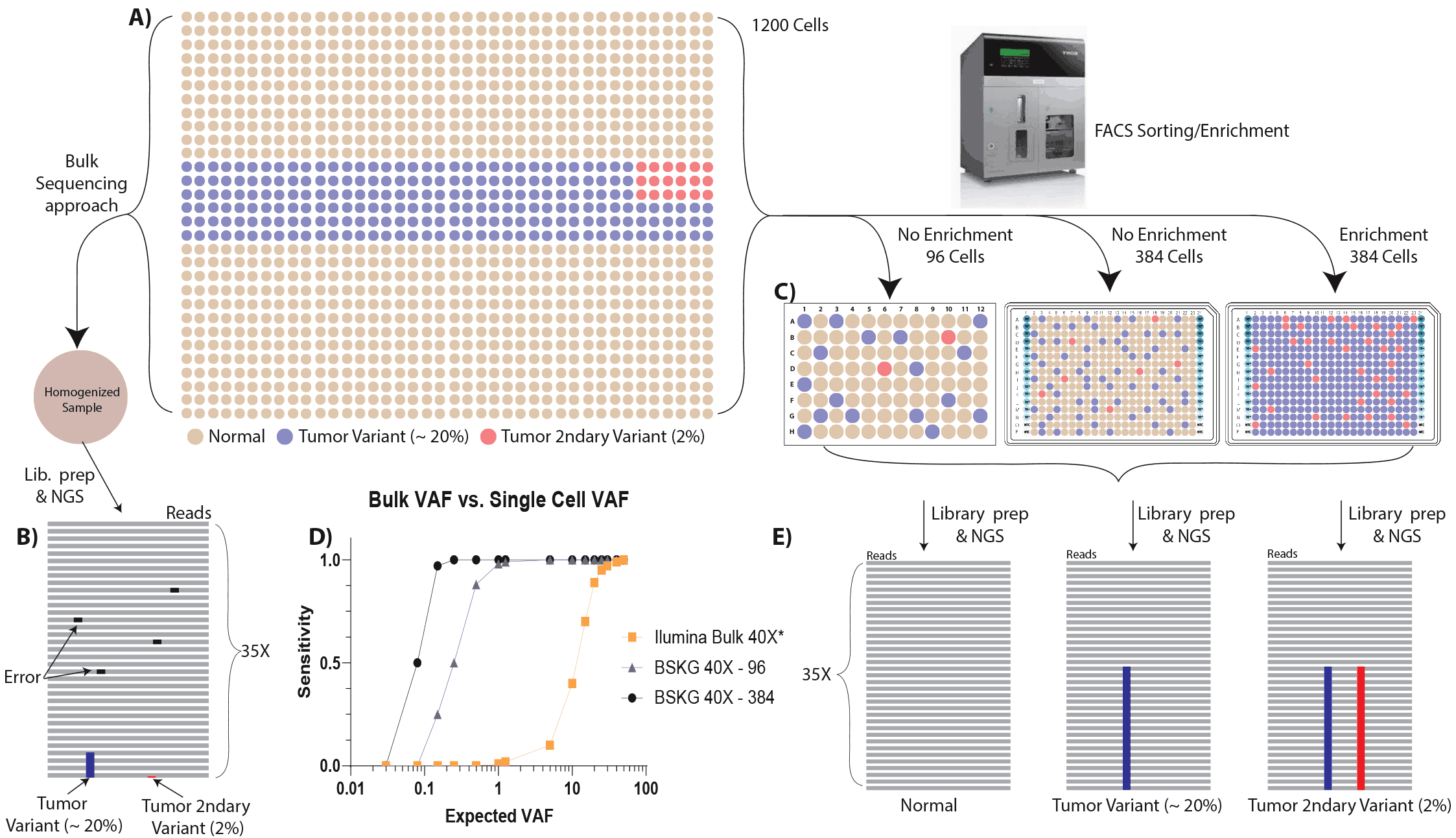
Figure 4. Single cell genome analysis increases Variant Allele Frequency (VAF) sensitivity. If 1200 cells (A) are analyzed which contain 20% tumor variant and 2% tumor secondary variant, by conventional bulk sequencing approaches, only the most prevalent tumor variants can be detected (B). This variant must be present in the majority of cells, while the tumor secondary variant cell is undetectable by sequencing of the sample homogenate. In contrast, the VAF sensitivity is increased by an order of magnitude by the single cell ResolveDNA platform. Using FACS sorting in combination with ResolveDNA single cell genome analysis the cells can be arrayed into a variety of microtiter plate formats for amplification, library preparation and sequencing. The data demonstrate, while the probability of capturing the target cells increases with cell throughput and enrichment, using non-enriched cells in a 96 rxn (or cell) format allows a dramatic improvement in SNV resolution. This is due to the binary presence or absence read-out (SC Heterozygous mutation VAF = 50% & SC homozygous mutation VAF = 100% or no mutation = 0). *https://www.illumina.com/content/dam/illumina-marketing/documents/products/whitepapers/whitepaper_wgs_tn_somatic_variant_calling.pdf.pdf&usg=AOvVaw2a27VPrqhhA1neYSZ5-k1m
(zero reads), a somatic variant (50% of reads) or a homozygous variant allele (100%) of reads. Even without enrichment, analyzing 96 cells from the tumor provide (Figure 4C) over an order of magnitude improvement in SNV resolution as measured by the relationship between VAF and SNV sensitivity(Figure 4D). Single cell VAF sensitivity increases with higher cell throughput and/or additively if a known cell marker is used to enrich the cancer cells prior to amplification, library preparation and sequencing analysis. Overall, single cell genome analysis unmasks both conserved, rare, as well as de novo mutations, enabling a impactful shift in SNV detection that largely defines tumor heterogeneity.
Cancer is not defined by a subset of genes. As VAFsensitivity increases, so will the number of genes associated with a pathology
Genetic diversity drives evolutionary processes. The modes of genomic mutation with pathological consequences are diverse, including activating or loss-of-function mutation, errors in DNA repair, dysfunction of tumor suppressor genes, and mutation that leads to improper programmed cell death. Due to the limitations in Variant Allele Frequency (VAF) detection sensitivity in tissue homogenates (referred to as "bulk"), only the most abundant gene variants (mutations) are detected. For de novo mutations, these variant mutations must be in greater than 50% of cells, which is a challenge given the typical purity of a tumor sample. Single cell genome sequencing increases gene mutation or VAF detection by orders of magnitude, which will uncover the less prevalent, but important variant alleles which may lead to new pathways to treat cancer more effectively.
While this initial study scope was limited in terms of cell numbers (~5 cells/group), the genomic diversity of the population was striking. First, all cells, both parental and resistant in this study displayed the expected ITD mutation in the FLT3 gene locus (Figure 7B). A notable difference in the FLT3 gene was our detection of a secondary, non-synonymous mutation (N841K) in the tyrosine kinase loop of the FLT3 gene of all resistant cells(Figure 7B). This mutation is clinically relevant and has been reported in AML patients9. To determine if this mutation was present in the parental population, a real-time genotyping qPCR assay was developed (Figure 7C). Using DNA from the populations the assay detected a weak, but present, signal from the naive cells. NTC samples displayed no amplification for either allele (data not shown). In contrast, the resistant population had
Data generated in an NGS Acute Myleiod Leukemia (AML) study (Figure 5) indicate that the use of extreme high depth sequencing (380x vs 35x) revealed ~ 11 times the number of variants7. While many of the emerging variants pre-existed in the population, nearly 20% were detected only after induction. The

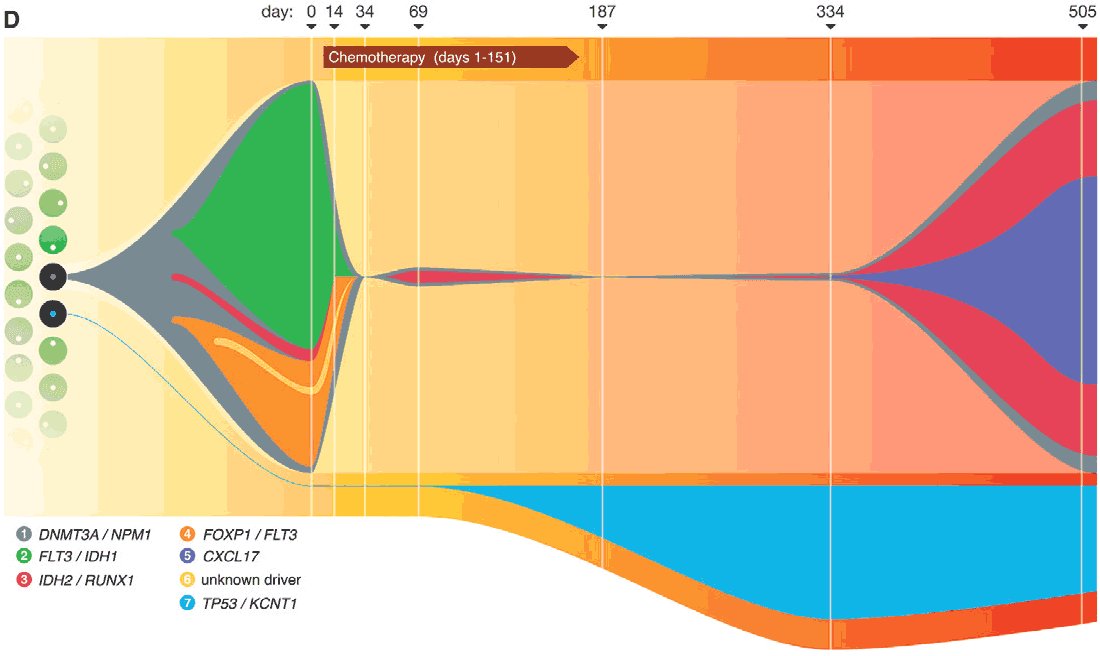
Figure 5. Clonal expansion of cancer. In this study, extreme high depth (380X) whole genome and whole exome sequencing of bulk tissues was employed to find newly emerging clones during treatment7. High depth and Single-cell sequencing allowed the stratification of 6 subclones that drove the disease over the independent origin of the TP53 mutant clonal population. Numbers in the legend refer to cluster assignments. The Chemotherapy label includes induction(day 1) and 4 rounds of consolidation chemotherapy at days 47, 81, 116, and 151.

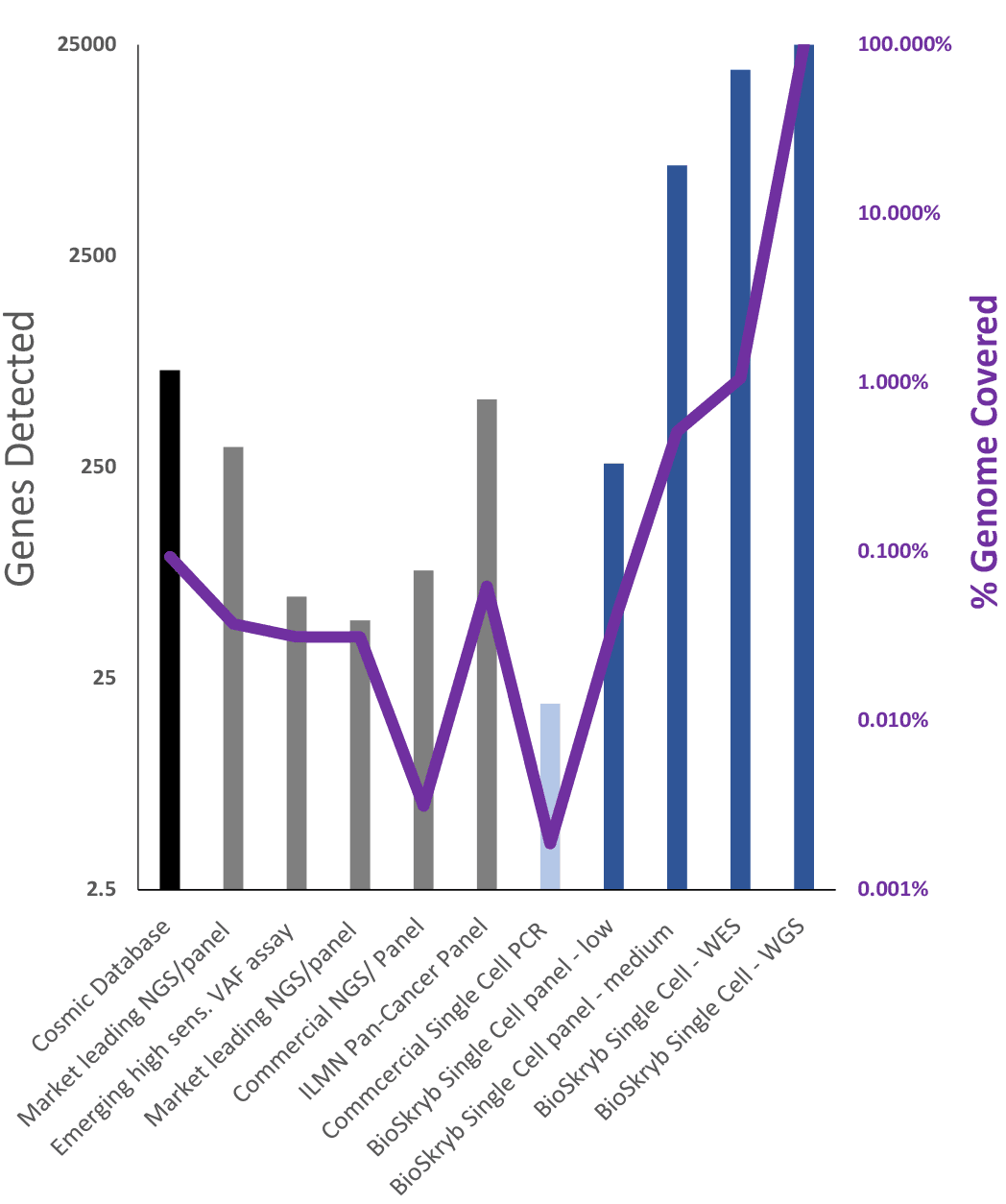
Figure 6. Comparison of genome sequencing panel technologies for both bulk and single cell analysis. NGS sequencing panels by definition cover only a portion of the genome. The COSMIC database has characterized over 700 genes related to cancer and many more mutations. The majority of commercial panels only cover a subset due to technical limitations (sensitivity) or limitations in multiplexing gene analysis (Commercial single cell PCR). ResolveDNA PTA-based genome amplification is compatible with small or medium panels, WES and WGS analysis, allowing a broad and complete range of discovery options.
use of single cell genome sequencing using PTA accomplishes the same increase in VAF sensitivity. However, for discovery, the use of limited gene panels will fall short of defining the clonal populations that lead to failed therapeutic approaches.
To enable greater tumor heterogeneity resolution the ResolveDNA system enables a wide range of sequencing options. After the amplification of a single cell genome, the amplified products can be used in down stream whole genome sequencing (WGS), Whole Exome Sequencing (WES) and targeted panel enrichment sequencing approaches. This enables high resolution single cell clonal fingerprinting of the population of single cancer cells. The high data quality data achieved by combining the ResolveDNA WGA and library preparation solutions with either WGS, WES or targeted panel enrichment (See enrichment protocols at BioSkryb.com) facilitates the ability to determine the lineage of a cancer cell population. Due to the high allelic balance generated with ResolveDNA PTA, resolution becomes only limited by the number of cells analyzed. Compared the existing panel-based analysis (Figure 6), the ability to delineate genome dynamics at high resolution allows discovery of the genetic heterogeneity of oncologic related pathologies.
Cancer is a highly complex state of pathology. The pathology

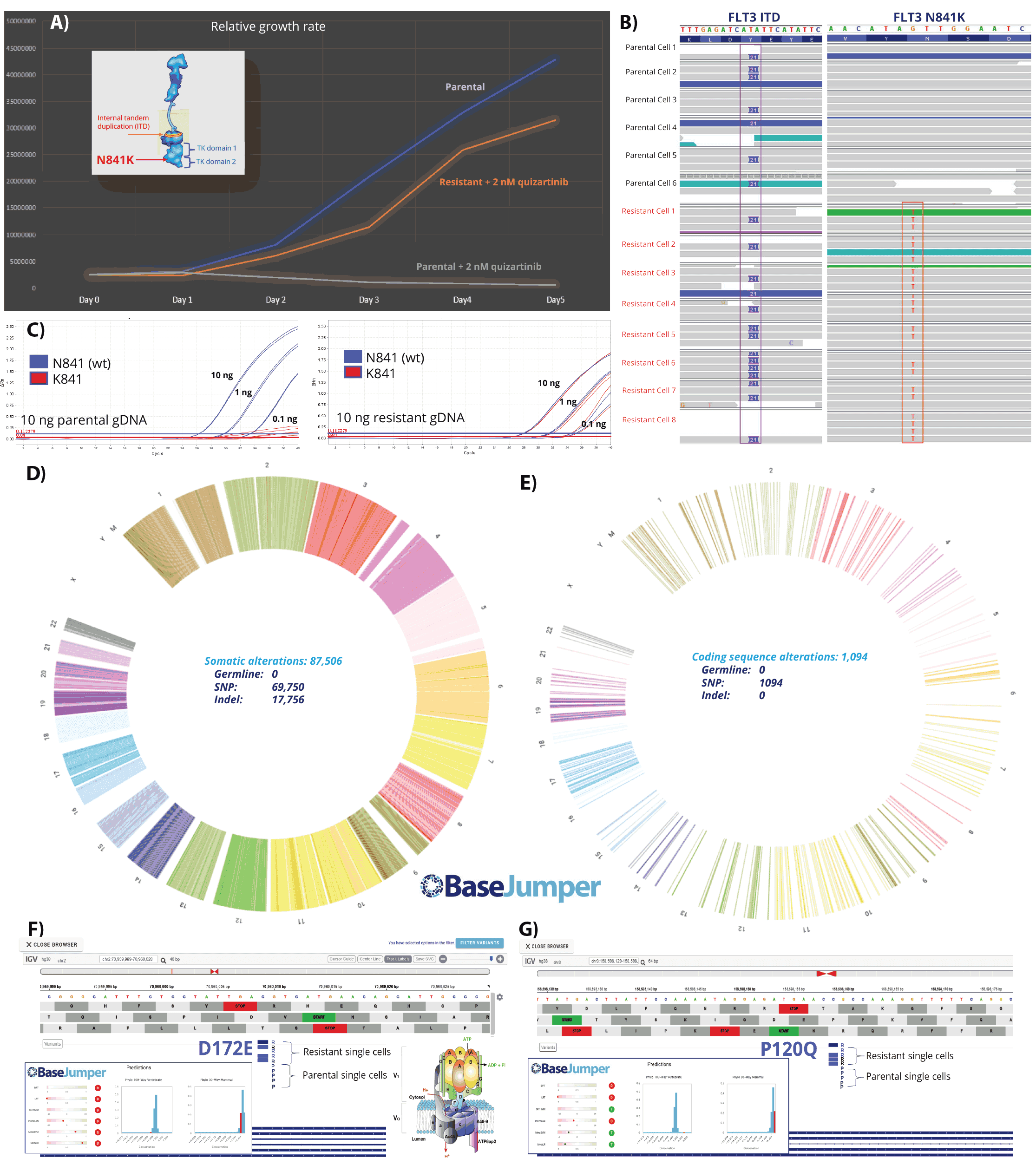
Figure 7. Clonal expansion of cancer. A patient-derived AML cell line, MOLM-13, was exposed to a tyrosine kinase inhibitor Quizartinib inducing resistance (A). Quizartinib targets the FLT3 kinase (A-inset). Both parental and resistant cells were sequenced (B) and found to harbor the FLT3 internal tandem duplication (FLT3-ITD), while the resistant cells (B) harbor an additional secondary mutation, N841K in the FLT3 gene. To determine if this clone existed in the parental population a qPCR method was developed to probe the bulk populations. This assay demonstrated the mutation was present at a low level in the parental cells, whereas in the resistant cells it was found in all cells (C). The sequencing data was then processed using BaseJumper™, which demonstrated nearly 88,000 additional variants. We then filtered these mutations to include only non-synonymous mutations in the exome and found ~ 1100 mutations. Two of these mutations, include an (F) drug efflux pump protein (v-ATPase) and a cell proliferation checkpoint protein (MLF-1) (G) both of which have been suggested to be associated with tumor progression in advancing AML. Both varaints were found only in a fraction of the resistant cells.
evolves over time and is influenced by the context of the tumor microenvironment. The environs of the tumor are affected by many intrinsic and extrinsic forces, one of which is the use of therapeutics in an effort to eradicate the specific disease.
In order to demonstrate the ability to discern these discrete clonal changes at single cell resolution, we performed a study designed to detect the modifications in the genome which occur during the exposure of a population of cancer cells to a therapeutic (Figure 7). We utilized a tyrosine kinase inhibitor (TKI) to target the FLT3 pathway, an important growth regulatory pathway in AML. Activating FLT3 mutations with constitutive kinase activity are common in AML, leading to uncontrolled cell proliferation8,9. To model this approach, we tested the ability to generate an additional new clonal subset of cells that became resistant to the drug over time. MOLM-13 cells are activated by the presence of an FLT3 internal tandem duplication (Figure 7A - inset). This duplication causes the FLT3 kinase to remain in an "on" kinase activity state which leads to an activation cascade of several downstream proteins (including PI3K, RAS, and STAT5) that cause the cells to continually divide. FLT-ITD induced AML is considered to result in poorer prognosis.
Upon exposure to the FLT3 inhibitor quizartinib, (Figure 7A) these cell populations initially declined in viability and growth rate. After several weeks of exposure a population of these cells became resistant to the drug and grew at normal rates compared to vehicle treated control. Both paternal and resistant were isolated as single cells by FACS into a series of 96 well plates. Single cell genomes from parental and resistant cells were then amplified using the ResolveDNA system for whole genome amplification. The amplified products were purified using the ResolveDNA bead system and subsequently converted to libraries for WGS using the ResolveDNA Library Preparation kit with the BioSkryb Genomics Library Preparation full length sequencing adapters. Libraries were first sequenced using an Illumina MiniSeq targeting 5 million reads/cell to assess quality. Cells passing quality metrics were submitted to high depth WGS targeting 600 million reads/cell. Upon completion of sequencing, BAM files were processed using the BioSkryb BaseJumper bioinformatic pipeline, down sampled (to 25X coverage) to 450 million reads/cell and resultant data was visualized on the cloud based platform.
While this initial study scope was limited in terms of cell numbers (~5 cells/group), the genomic diversity of the population was striking. First, all cells, both parental and resistant in this study displayed the expected ITD mutation in the FLT3 gene locus (Figure 7B). A notable difference in the FLT3 gene was our detection of a secondary, non-synonymous mutation (N841K) in the tyrosine kinase loop of the FLT3 gene of all resistant cells(Figure 7B). This mutation is clinically relevant and has been reported in AML patients9. To determine if this mutation was present in the parental population, a real-time genotyping qPCR assay was developed (Figure 7C). Using DNA from the populations the assay detected a weak, but present, signal from the naive cells. NTC samples displayed no amplification for either allele (data not shown). In contrast, the resistant population had
a clear heterozygous signal for the N841K secondary mutation (Figure 7C). Having identified this mutation, which is likely a chief contributor to the resistance mechanism of these cells, the data was further reviewed using the full breadth of the BaseJumper platform (Figure 7 D&E). Across all cells, a total of ~ 87,000 variants were found, approximately 75% of which were Single Nucleotide Variants (SNV), while 25% were indels. Using the BaseJumper platform the data was further filtered. We found ~1000 mutations that are predicted to result in a modification of a protein. Two of these non-synonymous changes were further examined (Figure 7 F&G). Neither mutation was found in every cell, nor did they appear to be co-related, suggesting they arose as separate clones. One of the variants (Figure 7F) ATP6V1B1, a vacuolar ATPase, is involved in regulating intracellular vesicle pH and extracellular pH and is required for the function of multidrug resistance pumps. It is regulated by Notch, Wnt, and mTOR signaling pathways. Given the well documented metabolism route of quizartinib by Cytochrome P450 3A and downstream phase II metabolism, which results in biliary excretion it is feasible that modifications in drug efflux could effect intracellular drug concentrations of the active AC866 metabolite that is the major species found after infusion.
A second non-synonymous modification was also studied further (Figure 7G). Myeloid Leukemia Factor 1 (MLF1) normally functions as a regulatory factor in the development of primitive hematopoietic cells and its deregulation contributes to hematopoietic dysplasia and leukemogenesis10. Myeloid leukemia factor 1 (MLF1) was first identified as the leukemic fusion protein NPM-MLF1 generated by the t(3;5)(q25.1;q34) chromosomal translocation. It has been shown that MLF1 induces p53 cell cycle arrest when cells commit to lineage during hematopoiesis. Expression of MLF1 is not restricted in hematopoietic cells11 and the aberrant over-expression of MLF1 is also reported in lung squamous cell carcinoma12 suggesting that MLF1 is involved in a common regulatory pathway related to tumorigenesis. Overall, MLF1 appears to have a regulatory control function in normal cells, where in contrast, overexpression is coordinated with pathology. Our findings of this mutant are consistent with the transformation. This nonsynonymous variant may be loss-of-function, which can no longer serve as a checkpoint in cell division. Moving forward, as more cells are assessed in this model, it will be critical not only to focus on these mutations but the collective pool of variants for additional insight into the mechanisms of resistance.
The variants discussed here in some case were in all of the cells of the respective parental or resistant population (FLT3-ITD, FLT3-N841K), and others were only found in a portion of cells (v-ATPase, MLF1). This variation created lineages which may drive the disease. An important aspect is the need to study these variations with the appropriate breadth. While single cell technologies do exist, most are limited to copy number variation detection or a small set of genes. This lack of resolution will inevitably limit the ability to dissect diseases involving clonal evolution. In this study, we found ~ 87K mutations between the cells, 1K of which were non-synonymous changes. While these variants require functional validation, they illustrate the

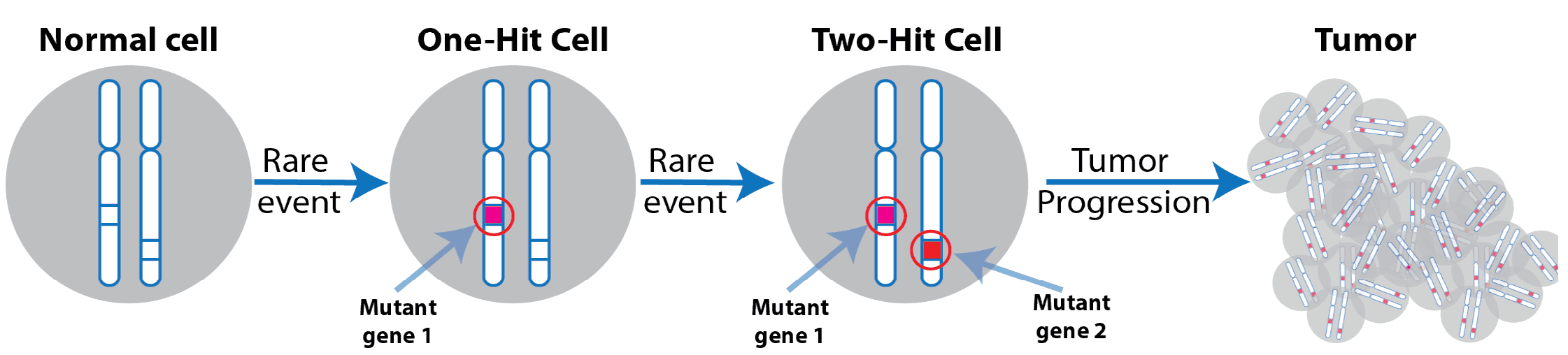
Figure 8. Two hit cancer hypothesis. The Knudson "two-hit" cancer hypothesis states that multiple mutations are required for the development of pathology. A normal cell first acquires a mutation either in the germ-line or as a somatic mutation, which pre-disposes the cell. A second event, either in the same gene or an alternative gene that allows the predisposed cells to advance and progress as a founding clone of cancer.
diversity of genomic variation in response to drug treatment and highlight the importance to comprehensively assess the variation. As more cells are processed these lineages based on mutation/variation will emerge. We will not find them if we don't look. Thus, BioSkryb Genomics highly recommends single cell whole genome amplification followed by whole genome sequencing or post amplification enrichment.
Co-occurrence of mutation drives disease. Evolutionary lineage results in distinct populations of cells that have a survival advantage and allow the disease to persist
It is has long been known that the development of acute pathologies such as cancer represent an evolution (Figure 8). In 1971 Knudson proposed the "two-hit" hypothesis and an underlying model of cancer development13. The hypothesis states that a single event creates a genetic pre-disposition or susceptibility to a given cancer. Later in time, an additional mutation occurs that allows the cells to grow uncontrollably. As proposed, this must occur at the individual cell level. This founding clone then begins to divide in an uncontrolled fashion, as it now has a fitness advantage.
The Knudson hypothesis (Figure 8), while controversial at the time is now widely accepted, particularly in hemotologic cancers. The sequencing of several AML tumors compared to healthy donors in the same age group reveals that the founding clone in AML may contain several gene variants. Within some models, only two gene mutations (JAK1 & PML-RARA) are required to initiate an aggressive Acute promyelocytic leukemia, whereas in human disease there appear to be up to 13 genetic variants involved in the progression of the tumor14.
However, these studies can only estimate the clonal structure. While the samples were deeply whole genome sequenced, they could not accurately delineate co-occurrence of mutation in a specific cell. In fact, this same group turned to the use of single cell methodologies to verify the predicted clonal structure of the tumor7 which revealed clonal predictions were not entirely accurate. Moreover, as the studies utilized tissue level sequencing, these rare cancer driving clones are not able to be detected unless sequenced approximately an order of magnitude deeper than standard recommended guidelines. In short, it is clear the resolution provided by single cell genome sequencing is beneficial. However, wide adoption of single-cell
genome sequencing has not yet occurred. The performance of the WGA chemistries have previously suffered from poor allelic imbalance, loss of coverage, and high error propagation limiting the ability to effectively analyze single cells genomes. ResolveDNA PTA now achieves the required uniformity, allelic balance and low error propagation required to achieve the required precision and sensitivity. This enables individual cell mutation co-occurrence analysis possible across the entire genome3.
The performance of the PTA-based ResolveDNA chemistry is critical as it allows highly sensitive detection of individual mutations within single cells. This allows the construction of

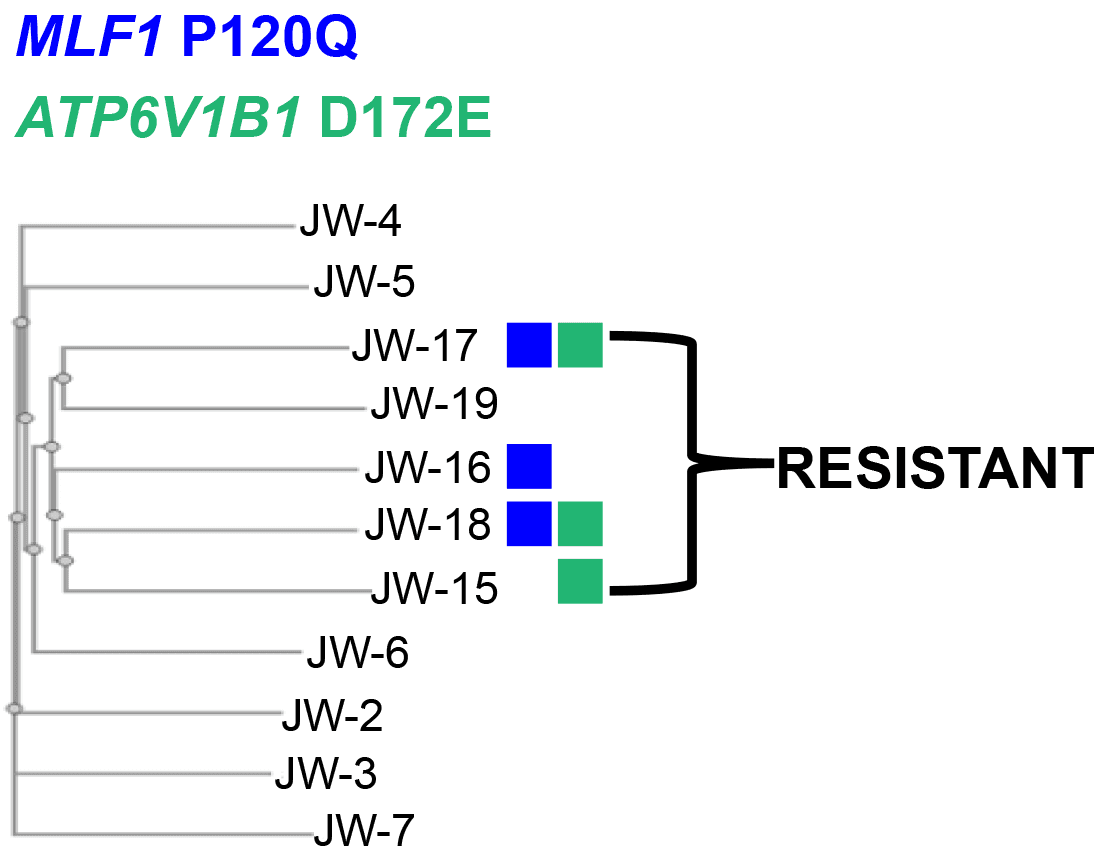
Figure 9. Co-occurrence drives population lineage. The ability to detect specific mutations with a single cell allows the creation of specific lineage trees. This lineage may ultimately make it possible to construct therapies to eradicate these populations. In this example clusters of resistant cells have a common ancestral FLT3 N841K mutation which was selected for during the cells exposure to Quizartinib. However, given the presence of a v-ATPase drug efflux mutation (ATP6V1B1 D1272E) these cells may have fitness advantage by removing the drug which inhibits the constitutive kinase activation. Thus, while we do not know whether D172E is activating or loss-of-function, one may consider directly targeting the v-ATPase with a drug like archazolid in conjuction with quizartinib as an initial follow-up study from these data. Fewer options are currently available for MLF1, however this is an active area of research in siRNA knockdown in lung cancer (Li, Min, et al 2018).
ancestral lineage trees. Because these lineage plots are driven by specific mutations (and other structural genomic changes) drug moieties targeting these sub-clones can be inferred for investigation. An example derived from the MOLM-13 experiment is described above (Figure 9). While the driver mutation in the lineage of the resistant cells appears to be the secondary N841K mutation to FLT3-ITD, several other potentially critical mutations exist as sub-clones. Importantly these subclonal mutation are involved in cycle cell arrest (MLF1) and drug export based resistance. The ability to resolve these alterations at single cell resolution provides options to determine an investigation path towards a remedy of a relapsed tumor.
Single-cell Tumor mutation burden (TMB) contributes to the evolution of the disease and how to target it for destruction
In cancer, as in any biological system, evolution results in positive selection of some variants and negative selection of other variants in a manner that depends on the overall selection pressure experienced by the population of cells. When the selective pressure induced by treatment or the host immune system increases the fitness of one subclone relative to another, the former subclone increases in frequency at the expense of the other. A requirement for positive selection of subclones in a tumor is that cells with a favorable combination of mutations must exist in the population in order for selection to occur.
Examples of low-frequency subclones being selected for following treatment of a primary tumor have been described. These include reported cases in non-small cell lung cancer and colorectal cancer15, and in HER2-positive breast cancer16.
Tumors with higher mutation burdens have a higher probability of containing existing variants that can be selected for following any treatment, allowing them to adapt to diverse treatment regimens. Consistent with this, tumor progression has been associated with increased clonal diversity17 and patient mortality risk has been linked to the presence of multiple subclones in a single tumor18. Although the increased genetic heterogeneity
affects patient outcomes, the exact nature of this relationship remains unclear.
The highly accurate single-cell variant profiles that are generated using ResolveDNA allow the measurement of tumor mutation burden at a resolution that is not feasible using bulk sequencing data (Figure 10). This single-cell readout of mutation burden enables the measurement of genetic heterogeneity of individual cells in a tumor, enabling estimation of the inherent selection potential of a tumor (Figure 10).
The use of tumor mutation burden as a treatment qualifier has gained traction in recent years. Accumulating evidence suggests that TMB may be a predictive biomarker of tumor response to immune checkpoint inhibitors (ICIs) in several cancer types19. The most robust initial responses to ICIs were observed in melanoma and non–small cell lung cancer (NSCLC), which typically have high mutation burden owing to the mutagenic effects of ultraviolet light and tobacco smoke, respectively19. Use of TMB is now a component of routine oncologic practice based on recent FDA approval of pembrolizumab for TMB-high solid tumors. The TMB-high subgroup was identified using a pre-specified cutoff of at least 10 mutations/MB, and issues remain regarding optimal cutoffs per tumor type based, in part, on baseline TMB distributions.
Prospective studies are needed for further evaluation and validation of the predictive utility of TMB in clinical practice. TMB has been shown to be a predictive biomarker for ICIs in both microsatellite instability-high (MSI-H) and microsatellite stable (MSS) cancers, and patient selection based on TMB levels may better select patients for ICIs or potentially expand the candidate pool for immunotherapy. Although current evidence indicates that TMB is associated with ICI efficacy, the mechanisms underlying the association between TMB and benefit from immunotherapy are incompletely understood. Assessing the mutation burden or rate of each cell may provide further benefit to resolve sub-populations of cells that can or should be alternatively targeted for therapeutic intervention.

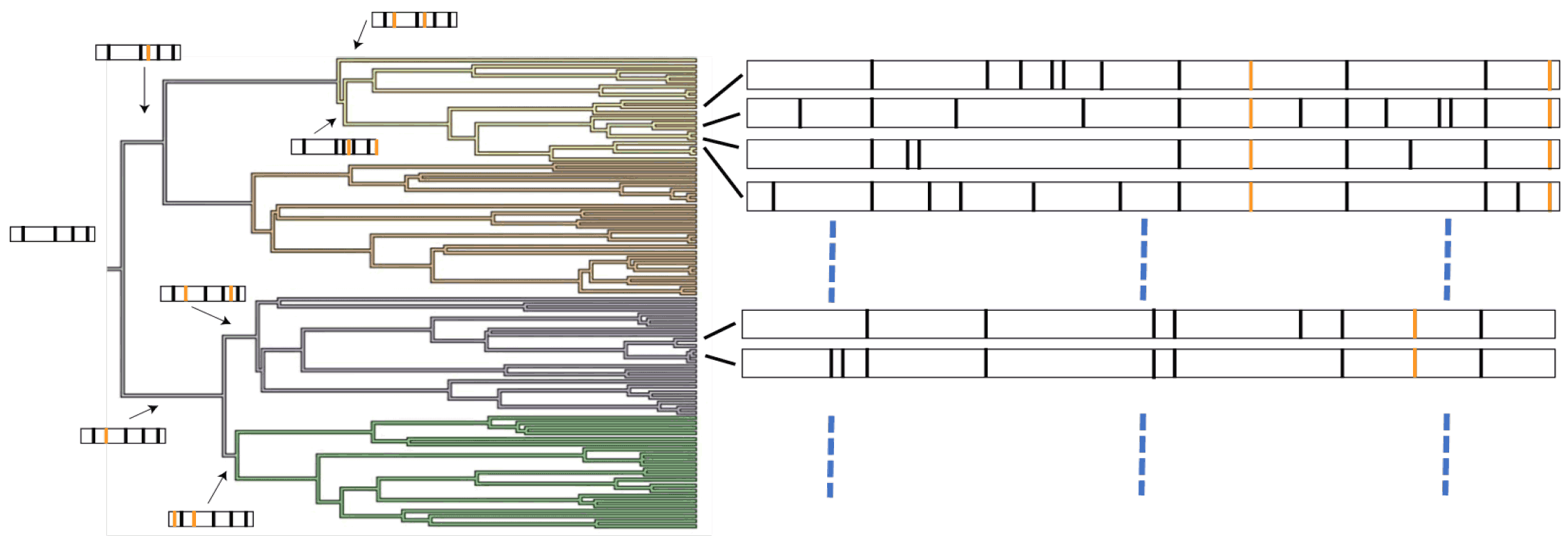
Figure 10. Tumor Mutation Burden Lineage: The branching structure details the clonal expansion which is directly linked the rate of mutation in a specific cell. The biology of tumorigeneis includes loss of tumor suppressor genes, dysfunctional DNA repair which can accelerate the number of mutations/cell. When sequenced as single cells, these can then be used to assess the TMB of each clone in the population.
The cancer genome is a function of SNV, CNV and translocation. Both structural and base-specific modifications contribute to disease pathology
Cancer is not comprised solely of single nucleotide point mutations. Many tumor types harbor some degree of structural variation including copy number variation or translocation. This was recognized with the ability to karotype a cell population. As an example, the MOLM-13 cells used in our studies are karyotypically atypical. They divide and multiply despite having multiple whole chromosome duplications and large deletions (Figure 11). The variation in chromosomal structure is strikingly variable between individual cells as judged by low depth sequencing analysis. To illustrate the degree of variability that may be present, we compared a known tetraploid breast cancer cell line SKBR3, to a control breast epithelial cell (Figure 12). Cultured SKBR3 cells were isolated as single cells using the BioSkryb Genomics FACS protocol, where a single cell was captured in each well of a 96 well plate. These cells were then frozen at -80o C, until ResolveDNA amplification was conducted. In this experiment, after purification, libraries were created using the Illumina DNA Prep library preparation kit. Libraries were then submitted for low pass sequencing on an Illumina MiniSeq, targeting 5 million reads/cell. Resultant BAM files were processed using the BioSkryb Genomics BaseJumper pipeline and further processed using Ginkgo20 for CNV visualization.
The degree of copy number variation in the cells is striking compared to a control normal breast primary single cell (Figure 12A) where no significant copy number changes are present. We then compared a sample of SKBR3 bulk DNA (Figure 12B)
to two individual SKBR3 cells (Figure 12 C&D). The diversity within the population was apparent. Compared to the bulk DNA (Figure 12B), the single cells from the same culture displayed alternative copy number variation in chromosome 1, 5, & 6. These cells harbored deletions(chromosome 8) in the isolated DNA as well as the single cells, although variation in the deletion segments is clearly apparent between the two. The changes are not insignificant, where each (500kb) window represents the amplification or deletion of a multitude of genes. Given the variability, it is clear from copy number variation an immortalized cell line can not only survive but grow aggressively in culture. We have found these cell models are very helpful in developing the ability to define the required parameters to analyze both copy number and single nucleotide variation at single cell resolution.

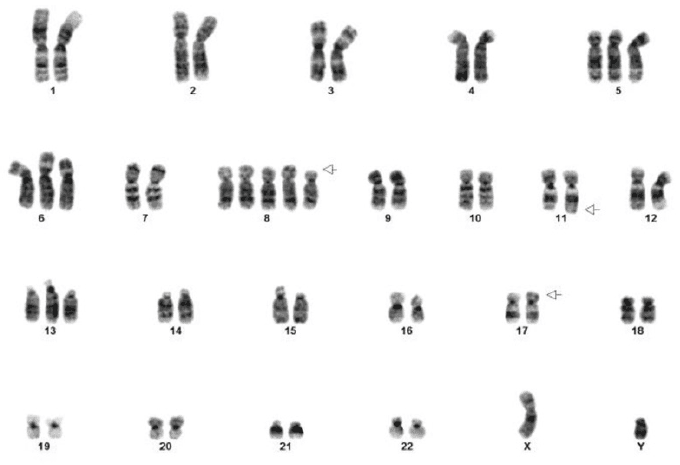
Figure 11. MOLM-13 AML Karyotype. Cancer cells often have surprising degrees of aneuploidy. This patient-derived cell line contains a chromosome 8 pentaploidy a trisomy at chromosome 5, 6, & 13 and deletion of chromosome 11 & 17.

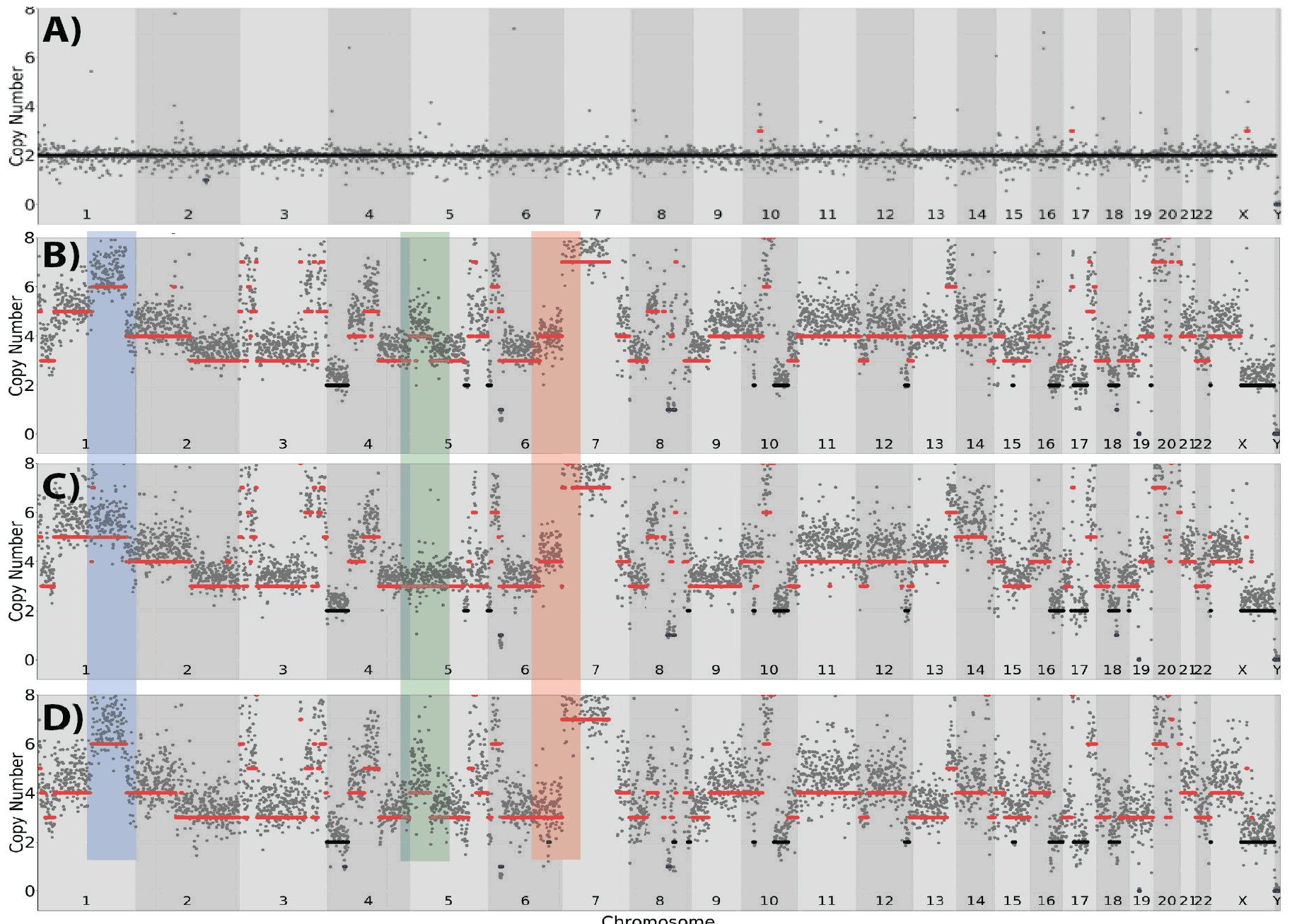
Figure 12. SKBR3 Singlecell copy number analysis. Compared to a normal primary single breast epithelial cell (A) where the chromosome structure appears to be intact and wholly diploid the bulk DNA from a group of SKBR3 cells (B) had a high degree of copy number variation (CNV). Similar to the pattern in the bulk DNA, dramatic CNV variability was observed in single cells from the same culture. However, clear differences (blue highlight) at the single cell level were apparent. In one single cell (D) the CNV pattern of Chromosome 1 was similar to the bulk DNA while in another single cell (C) the degree of duplication is less compared to other cells and the bulk DNA. Similar patterns of differential CNV patterns were in chromosomes 5(green highlight) and 6 (red highlight).
stages of ductal carcinoma in situ/invasive ductal carcinoma(DCIS/IDC). A portion of the samples are first dissociated, then cryo-preserved. Other portions of the samples are preserved in formalin for standard histopathology and IHC analysis. To conduct single-cell analysis the cells were carefully thawed and resuspended. A portion of the cell suspension was used to assess tumor burden (by IHC) and the remaining sample was incubated with an EpCam primary fluorophore-conjugated antibody which was subsequently used for FACS single cell isolation (Sony SH800) in 96 well plates, immediately centrifuged and placed on dry-ice. Single cells were stored at -80C until amplification, library preparation and sequencing. Each sample generated ~10 plates for downstream analysis. Amplification and purification were conducted using the standard ResolveDNA protocols for WGA and bead clean-up. For these samples, Illumina DNA Prep(tagmentation-based) was used for library preparation which

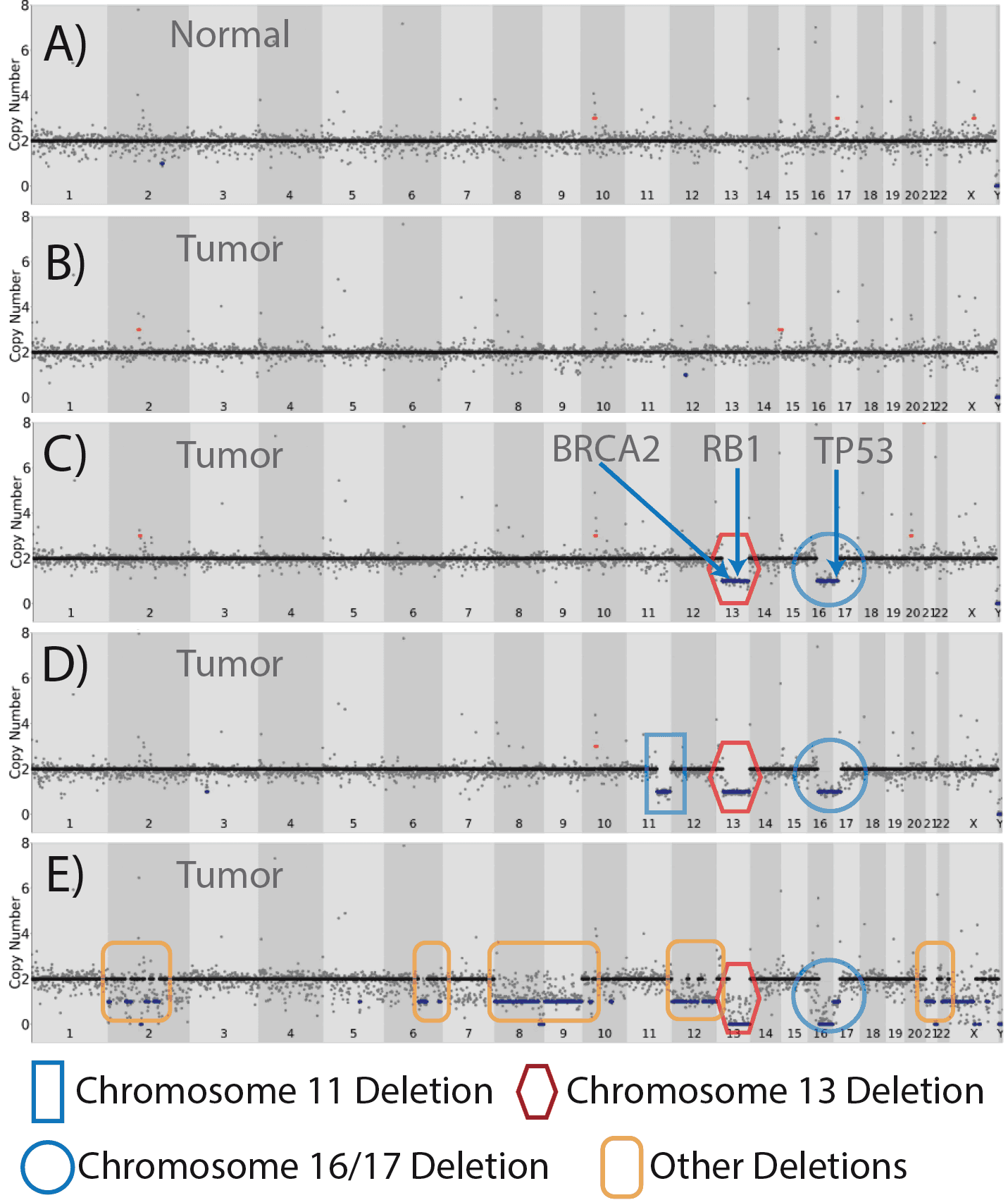
Figure 13. CNV analysis of a clinically resected DCIS breast tumor. Compared to a normal breast epithelial cell (A) where the chromosome structure to be intact with two alleles at each chromosome, CNV profiles from a single tumor sample were diverse. While normal CNV profiles were found in the tumor sample (B) three additional CNV profiles were found within this sample of 31 cells (C-E). One profile contained deletions in chromosome 13, 16 and 17, which represented ~ 20% of the cells (C), while a second dominant profile (~ 25% of cells) had an additional deletion in chromosome 11 (D) Finally one cell was found to have a massive set of deletions (E) in chromosomes 2, 6, 8, 9, 12 13, 16, and 17.
was quality evaluated by low pass sequencing prior to high depth sequencing using a NovaSeq 6000 S4. Fastq and BAM files were processed using the BioSkryb Genomics BaseJumper cloud based BioInformatics platform and subsequently visualized to defined SNV. A custom script was developed using Ginkgo to assess CNV using the low pass NGS QC data.
We first assessed the performance of the chemistry for the samples. We found the genome mapping and coverage exceeded 95% in all but two of ~30 samples, with low percentage detection of the mitochondrial genome. Further details of the performance can be found in the following Illumina/BioSkryb Application note21.
The samples revealed significant diversity in terms of copy number variation (Figure 13). Samples recovered from the non-tumor site had normal CNV (Figure 13A), with minimal chromosomal duplication, amplification or deletion. In contrast, samples biopsied from the tumor site had significant diversity in terms of copy number variation. While some of the cells from the tumor biopsy appeared to have normal CNV patterns (Figure 13B), we saw three additional patterns of CNV. The first pattern (Figure 13C) was a deletion in chromosomes 13, 16, and partially from 17. Given that BRCA2 and RB1 are located on chromosome 13, loss of both suppressor genes indicated that this clone may have impaired DNA repair and loss of control in terms of proliferation. The partial deletion of Chromosome 16 is a common finding but not fully understood modification in breast cancer. In addition, the partial deletion of chromosome 17, nearly the entire p arm, includes a deletion of TP53 (p53), but does not appear to include BRCA1, located on the q arm of the chromosome, which appears to be intact. This clone, based on CNV, represented approximately 20% of the cells that were sequenced. A second CNV variant was also found, which included an additional deletion of the long arm of chromosome 11. This deletion has been proposed to be associated as part of the progression from DCIS to invasive cancer22. It is notable that this clone also contained the deletion in 13, 16 and 17 and represented ~ 24% of the cells sequenced. A third, more rare differential pattern of CNV (Figure 13E) was found in one cell (out of 30 cells sequenced ~ 4%). We noted additional deletions in chromosome 2, 6, 8, 9, 12, 22 with near complete deletions in 13 and 16, a portion of 22, and a partial deletion in 17 (similar to other clones), but this cell did not contain a deletion in chromosome 11. Indeed this may be a cell that is dying or unhealthy.

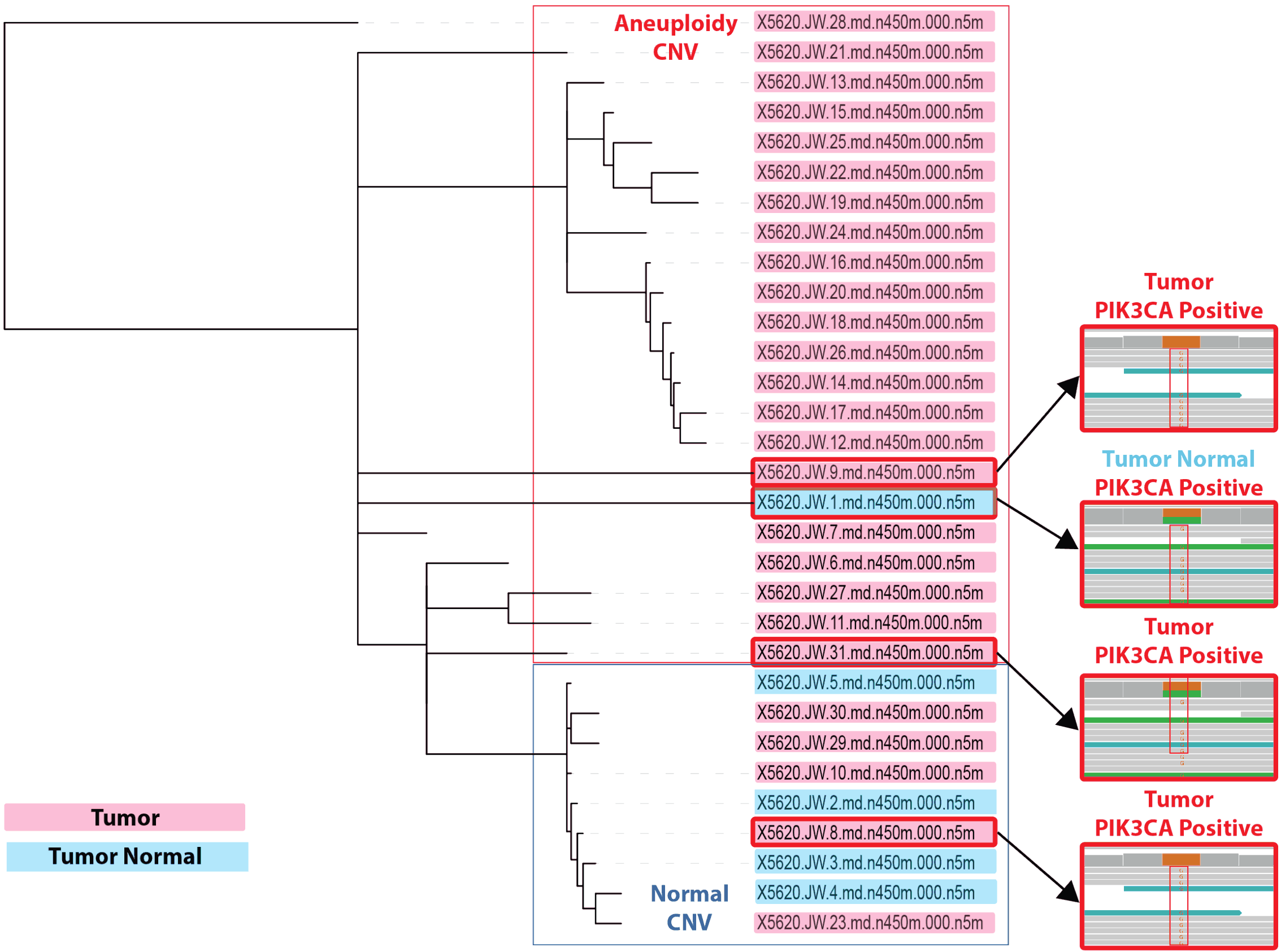
Figure 14. PIK3CA mutation Analysis within the CNV context. PIK3CA H1047R mutations were found in several cells within the tumor. However, it appears the copy number status was differentiated between the tumor cells, where cells that harbored the H1047R mutation had normal CNV patterns. In addition, we found a single cell within the normal non-tumor sample which also contained PIK3CA H1047R, which was resected ipsilaterally from the same breast. Overall, of the 31 cells sorted in this experiment, based on the dendrogram and PIK3CA H1047R mutations, a significant number of the cells had an alternative mutation pattern and/or a differentiated CNV profile.
Given the findings that aberration in chromosome 8, 9, and 12 structure are common in breast cancer, typically involved in the advancement of the disease, the results are plausible as an indication of these lesions contributing to the advancement of this tumor.
While analysis of these samples continues, we used the deep sequencing data to assess the degree of single nucleotide variants in addition to the copy number variation. We identified an important oncogenic mutation in the gene PIK3CA. Phosphatidylinositol 3-kinases (PI3Ks) are a group of lipid kinases that regulate signaling pathways involved in cell proliferation, adhesion, survival and motility. The PI3K pathway is considered to play an important role in tumorigenesis. Activating mutations of the p110α subunit of PI3K (PIK3CA) have been identified in a broad spectrum of tumors. Analyses of PIK3CA mutations reveals that they increase the PI3K signal, stimulate downstream Akt signaling, promote growth factor-independent growth and increase cell invasion and metastasis23. PIK3CA gene mutations are linked to breast cancer, as well as to cancers of the ovary lung, stomach, and brain.
High depth sequencing revealed several samples that contained the PIK3CA H1047R mutation (Figure 14). This mutation was found in approximately 10% of the tumor samples. We further noted that patterns of CNV and PIK3CA mutation were independent. In fact, the tumor cells with deletions of tumor suppressor genes (Figure 13 C,D,E) were devoid of the PIK3CA mutation, while cells that contained the PIK3CA mutation either had normal CNV profiles or a minor degree of aneuploidy. Notably, was the detection of the PIK3CA H1047R mutation within the normal biopsy sample. While the tumor normal sample was biopsied from the same breast, it was distal of the resection site. One possibility is the resection margin did not capture all of the cells that harbor the PIK3CA H1047R mutation, while we noted the majority of normal biospy single cells samples appeared to have normal CNV profiles. It is possible the increase in sensitivity from the single-cell genome method has detected a pre-malignant event.
Taken together, the data indicate that the CNV patterns in this sample are highly diverse. The genes amongst these chromosome deletions are consistent published patterns of breast tumors. Combined with the single nucleotide variation data the diversity of this tumor is significant. In some samples, it appears the tumor cells will be driven by the PIK3CA mutation, whereas other samples will likely be driven by the deletion of tumor suppressor genes. Even using a small number of cells, single-cell analysis has provided evolutionary insight into the tumor. Further efforts with this patient data will include the use of BaseJumper to map additional mutation profiles onto the variable CNV profiles. This may yield possible drugable targets for the cells that are driven by their aneuploidy state, which are otherwise considered not treatable. In addition, the discovery of new markers and protein coding alterations not previously annotated may yield new targets and/or biomarkers. We posit that mutations and structural variation in the intronic and intergenic regions of the genome offer a wide expanse of new therapeutic targets. Accordingly, we believe that whole genome sequencing, while costly, will provide the most optimal long term value from high precision single cell sequencing analysis from precious patient samples. The challenge we all recognize is generating the richest data on enough individual cells within these highly heterogeneous tumors to allow accurate diagnosis and development of personalized therapeutic approaches. Such data may be utilized by the institutional tumor boards across the spectrum of cancer centers globally.
High resolution single cancer cell genome analysis using PTA will revolutionize therapeutic approaches
While cancer treatment over past several decades has improved dramatically, so has the incidence rate. Currently, nearly 15 million people worldwide are diagnosed every year. In many aspects, the effective treatments have been elusive. Our ability to improve patient outcomes is dependent on high definition and high resolution analysis of pathology. This increase in resolution requires improvement in data processing, visualization and interpretation, which by definition is complex.
At BioSkryb Genomics, there is clear understanding and acknowledgment that bioinformatic processing of the data is critical and the underpinning of improved decision making processes. Moreover, while the primary focus of this application note has focused on the genome structure, further developments to fully understand the multi-modal nature of the genome regulation and output in terms of transcription and translation are important factors influencing discovery, drug development and potentially new therapeutic constructs and combinations (Figure 15). Given the exquisite control and flexibility of Primary Template-directed Amplification, we at BioSkryb Genomics are enthusiastically pursuing these advanced applications. These advancements will be centralized by SNV, CNV and mutation burden that shape the biology of tumors and their ability to survive under intrinsic and extrinsic selective pressures. Discovery of the genomic diversity can and should begin with the routine process of single cell isolation, genome amplification and next generation sequencing analysis.

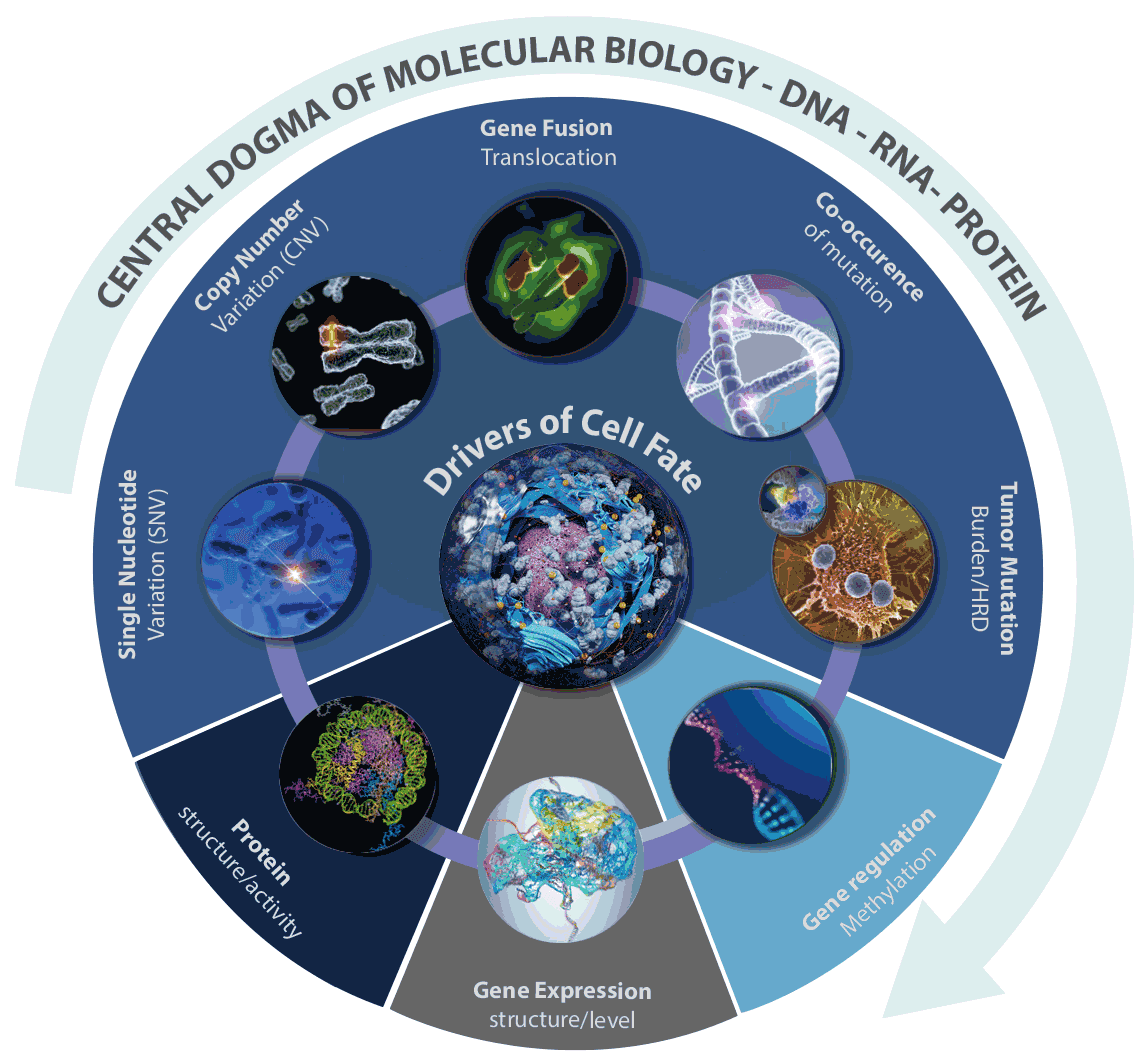
Figure 15. Factors affecting tumor heterogeneity. Several molecular biology states contribute to cellular heterogeneity, which drive evolution and survival of the tumor cells. These include, single-cell Variant Allele Frequency(scVAF), copy number changes (scCNV), tumor mutation burden (scTMB), cytosine methylation, as well as transcriptional, translational and histone modification contributions.
References:
- Nowell, P.C., The clonal evolution of tumor cell populations. Science, 1976. 194(4260): p. 23-8.
- Mendelsohn, J., et al., The Molecular Basis of Cancer, 4th Edition. 4th ed. 2015, Philadelphia, PA: Saunders/Elsevier. 888. line-height:normal;mso-outline-level:1"> The Molecular Basis of Cancer, 4th Edition. 4th ed. 2015, Philadelphia, PA: Saunders/Elsevier. 888.
- Gonzalez-Pena, V., et al., Accurate genomic variant detection in single cells with primary template-directed amplification. Proc Natl Acad Sci U S A, 2021. 118(24).
- BioSkryb Genomics, i., ResolveDNA Whole Genome Amplification Kit For high-quality single-cell and low-input DNA amplification, in www.bioskryb.com, B. Genomics, Editor. 2021, BioSkryb Genomics: Durham, NC. USA.
- Wooster, R., et al., Identification of the breast cancer susceptibility gene BRCA2. Nature, 1995. 378(6559): p. 789-92.
- Dagogo-Jack, I. and A.T. Shaw, Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol, 2018. 15(2): p. 81-94.
- Griffith, M., et al., Optimizing cancer genome sequencing and analysis. Cell Syst, 2015. 1(3): p. 210-223.
- Park, I.K., et al., Receptor tyrosine kinase Axl is required for resistance of leukemic cells to FLT3-targeted therapy in acute myeloid leukemia. Leukemia, 2015. 29(12): p. 2382-9.
- Matsuno, N., et al., A novel FLT3 activation loop mutation N841K in acute myeloblastic leukemia. Leukemia, 2005. 19(3): p. 480-1.
- Zhu, C., Y. Wei, and X. Wei, AXL receptor tyrosine kinase as a promising anti-cancer approach: functions, molecular mechanisms and clinical applications. Mol Cancer, 2019. 18(1): p. 153.
- Yoneda-Kato, N., et al., The t(3;5)(q25.1;q34) of myelodysplastic syndrome and acute myeloid leukemia produces a novel fusion gene, NPMMLF1. Oncogene, 1996. 12(2): p. 265-75.
- Sun, W., et al., Identification of differentially expressed genes in human lung squamous cell carcinoma using suppression subtractive hybridization. Cancer Lett, 2004. 212(1): p. 83-93.
- Knudson, A.G., Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A, 1971. 68(4): p. 820-3.
- Welch, J.S., et al., The origin and evolution of mutations in acute myeloid leukemia. Cell, 2012. 150(2): p. 264-78.
- Hiley, C., et al., Deciphering intratumor heterogeneity and temporal acquisition of driver events to refine precision medicine. Genome Biol, 2014. 15(8): p. 453.
- Janiszewska, M., et al., In situ single-cell analysis identifies heterogeneity for PIK3CA mutation and HER2 amplification in HER2-positive breast cancer. Nat Genet, 2015. 47(10): p. 1212-9.
- Birkbak, N.J. and N. McGranahan, Cancer Genome Evolutionary Trajectories in Metastasis. Cancer Cell, 2020. 37(1): p. 8-19.
- Andor, N., et al., Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat Med, 2016. 22(1): p. 105-13.
- Sha, D., et al., Tumor Mutational Burden as a Predictive Biomarker in Solid Tumors. Cancer Discov, 2020. 10(12): p. 1808-1825.
- Garvin, T., et al., Interactive analysis and assessment of single-cell copy-number variations. Nat Methods, 2015. 12(11): p. 1058-60.
- Zawistowski, J., et al., Single-cell oncogenic mechanistic heterogeneity defined by PTA in primary Ductal Carcinoma In Situ, in Application note, B. Genomics, Editor. 2021, BioSkryb Genomics: www.bioskryb.com. p. 1-5.
- Newsham, I.F., The long and short of chromosome 11 in breast cancer. Am J Pathol, 1998. 153(1): p. 5-9.
- Ligresti, G., et al., PIK3CA mutations in human solid tumors: role in sensitivity to various therapeutic approaches. Cell Cycle, 2009. 8(9): p.1352-8.


For more information or technical assistance: info@bioskryb.com
TAS.014, 01/2022